ОБЪЕДИНЕНИЕ КОМБУСТИОЛОГОВ
«МИР БЕЗ ОЖОГОВ»
Адаптация к ожоговой травме: проблемы и перспективы
Ушакова Т.А.
ФГУ «Институт хирургии им.А.В.Вишневского Росмедтехнологий», Ожоговый центр, Москва.
Актуальность проблемы.
Современная медицина располагает большим арсеналом средств и методов для оказания квалифицированной помощи тяжелообожженным пострадавшим. Разработаны основные принципы выведения пациента из шоковой гиповолемии, нутритивной поддержки, деэскалационной антибиотикотерапии, раннего хирургического лечения. В последние годы в ведущих ожоговых клиниках страны все шире применяют высокотехнологичные способы экстракорпоральной детоксикации и иммуномодуляции.
Однако, несмотря на достигнутые успехи, при обширных и глубоких ожогах сохраняется высокая частота развития полиорганной недостаточности (ПОН) и сепсиса, летальность при которых имеет тенденцию к возрастанию (Саркисов Д.С., 1980; Алексеев А.А., 1989, 1993; Крутиков М.Г., 2004).
По данным института им. Н.В. Склифосовского у 94% погибших от ожогов имелись морфологические признаки ПОН и у 48% — сепсиса (Смирнов С.В. с соавт., 1999). Результаты исследований, проведенных в Институте им. А.В. Вишневского, демонстрируют зависимость частоты развития сепсиса от наличия симптомов синдрома системного воспалительного ответа (СВО): при ССВО-3 сепсис диагносцирован в 68.57% случаев, летальность при этом составила 34.29%; при ССВО-4 — 95.24% и 47.6% соответственно (Крутиков М.Г., 2005).
Высокие показатели летальности (в среднем около 30%) превращают сепсис в общемедицинскую проблему (Артамонов Р.Г., 2006). Подобная статистика характерна для всех развитых стран Европы и Северной Америки, где при лечении септических больных в отделениях реанимации и интенсивной терапии широко используют самые современные антибиотики. Еще более высокий (от 40 до 80-90%) уровень летальности констатируется при септическом шоке, при этом на него мало влияют достижения антибактериальной химиотерапии и мероприятия по санации очагов инфекции (Артамонов Р.Г., 2006). В США из 750 000 случаев зарегистрированного сепсиса (из них 9% тяжелой формы и 3% септического шока) отмечено 210 000 летальных исходов (Christ–Crain M at Muller B, 2007).
Сам факт обреченности трети пациентов с диагносцированным сепсисом и увеличение вероятности летального исхода в два раза при появлении признаков ПОН свидетельствует либо о фатальности данного состояния, либо о неправомочности принципов и приоритетов проводимого лечения (Козлов В.К., 2006).
Фатальная обреченность пациентов с ПОН и сепсисом при термической травме и отсутствие тенденции к снижению летальности в условиях современного комплексного лечения с использованием высокотехнологичных методов предполагают к поиску альтернативных способов решения данной проблемы. Необходимо отметить, что проблема эта сохранила высокую актуальность и обрела междисциплинарную значимость, однако при этом путь к радикальному перелому ситуации становится все более тернистым и трудоемким (И.А. Ерюхин, 2003).
Введение в проблему
Метаболические проблемы у ожоговых пациентов могут появиться в любое время: от немедленного послеожогового периода до позднего – выздоровления. Их отличают частота, тяжесть и продолжительность (Pruitt BA at al, 1985; Atiyeh BS at al, 2000). Различают ранние (немедленные послеожоговые и постресусистентные) и более поздние метаболические нарушения.
В 30-гг. прошлого века Cuthbertson at al. (1932) составили схему метаболических ответов больных на повреждение, актуальную, на наш взгляд, до сих пор.
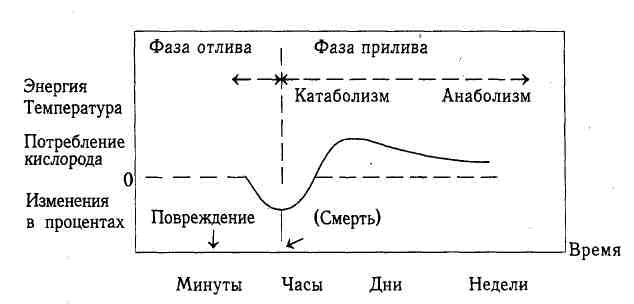
Схема метаболического ответа на повреждение
по Cuthbertson et al., 1932 (Pruitt BA et al, 1985)
Авторы этой модели выделяют по времени фазы отлива и прилива. Фаза отлива соответствует немедленным последствиям повреждения, которые уменьшают энергетические затраты в покое. В этот период отмечаются уменьшение объема циркулирующей плазмы (после тяжелого ожога) и потеря эффективного циркулирующего объема (потери в так называемое третье пространство, возникающее в результате травмирования тканей). Эти изменения вызывают резкие нарушения микроциркуляции, агрегацию и секвестрирование клеток крови (сладж-синдром), последующую гиперкалиемию и гипонатриемию, обусловленную выходом натрия из сосудистого русла вместе с жидкостью.
Компенсаторные на снижение объема циркулирующей плазмы гемодинамические изменения со стороны организма, заключающиеся в централизации кровообращения и периферической вазоконстрикции, усугубляют системную ишемию, а развивающиеся гипоксия и ацидоз вызывают депрессию функциональной активности центральных органов и систем. Таким образом, создается патофизиологическая основа для ранней полиорганной недостаточности (ПОН), которая является негативным последствием, как травмы, так и адаптивных реакций. Необходимо отметить, что подтверждением ранней ПОН служат и результаты молекулярно-биохимических исследований, интенсивно развивающихся в последние годы, о чем будет подробно изложено ниже.
Известно, что ранние метаболические проблемы, связанные с гипоксией, коагулогическими сдвигами, серьезными водно-электролитными нарушениями и дисбалансом кислотно-основного состояния в сторону ацидоза, давно и успешно решаются.
Однако, осознания важности сохранения у пострадавшего умеренного функционального напряжения всех органов и систем, от которого зависит успешный выход из длительного состояния повреждения, на наш взгляд, не произошло, ибо в интенсивном лечении этого шокового периода мы стремимся к приведению всех измененных показателей к норме, что находится в противоречии с адаптивным подходом к процессу восстановления после травмы. Нивелирование ответной реакции организма уже на ранних стадиях лечения приводит в дальнейшем к необходимости повторного ее запуска (повреждение-то остается!) и лежит в основе раннего метаболического истощения. О дополнительной коррекции ранней ПОН речь не идет вовсе.
Метаболические проблемы постресусистентного периода связаны со многими причинами. Во-первых, фаза прилива по Катбертсон соответствует периоду реперфузии (по современной терминологии) со всеми вытекающими последствиями проникновения в кровь клеточных дериватов и микробных продуктов в виде выраженной интоксикации и усиления обменных процессов. Во-вторых, после периода дегидратации в организме происходит задержка жидкости и натрия на фоне гипокалиемии. Однако, в связи с повышением проницаемости капилляров, а также использованием в шоке сверхобъемов, жидкость задерживается в экстрасосудистом пространстве, инкриминируя раннюю постресусистентную легочную отечность и интерстициальную отечность всех органов и систем.
В то же время, сохраняющаяся в связи с этим дегидратация стимулирует ренин-ангиотензиновую систему в почках, вызывая компенсаторное повышение активности альдостерона с целью задержки натрия (развивается гипернатриемия) и воды, которая вновь устремляется в третье пространство. Возникает вторичный гиперальдостеронизм, корригировать который подчас не удается. Попытки устранения дегидратации и гипернатриемии дополнительными объемами жидкости создают порочный круг: эта жидкость вновь поступает в интерстиций, а натрий остается в сосудистом русле.
Т.е., водно-электролитные нарушения этого периода также связаны как с самим поражением, так и с последствиями интенсивного лечения: не только стремлением к нормализации параметров в шоке, но и ведением пациента на длительной гемодилюции. В результате, исподволь развивающаяся ПОН, проявляется уже клинико-лабораторными свидетельствами системного застоя, который способствует еще большему нарушению клеточного метаболизма. Происходит дальнейшее угнетение ответной реакции организма, чреватое срывом адаптации.
Таким образом, классическое патогенетическое восприятие демонстрирует набор данных, свидетельствующих о нарушениях в организме и нуждающихся в приведении к норме; а адаптивное восприятие развития ответной реакции - выделение адаптивных (защитных, компенсаторных) и дезадаптивных реакций, поддержка первых на оптимальном уровне и коррекция вторых.
Актуальность адаптивного подхода в коррекции метаболизма у пострадавших с тяжелой ожоговой травмой и актуальность акцентирования внимания на развитии ранней ПОН обусловлена, на наш взгляд, непосредственной связью синдрома ПОН с дальнейшим развитием сепсиса.
Широко распространенное мнение о первичности сепсиса в этиологии ПОН, а о ПОН как основном патогенетическом факторе в развитии его стадий, вносит некоторую двойственность в проблему сепсиса.
На основании современной классификации системного воспалительного ответа (СВО), принятой на Калужской согласительной конференции в 2004 году, пострадавших с ожоговой болезнью с легкостью можно отнести к пациентам с сепсисом: имеется первичный инфекционный очаг и системная реакция организма. А всем пациентам с имеющимися признаками органной дисфункции на фоне системной воспалительной реакции необходимо ставить диагноз тяжелого сепсиса. Однако, конкретизация терминологии, представленной также и в работе И.А. Ерюхина и С.А. Шляпникова (2003) о стандартах в хирургии, заставляет вновь поднять этот вопрос.
Дело в том, что согласно рекомендациям Калужской конференции под органной дисфункцией понимается не что иное, как клинически выраженная недостаточность органов и систем. На это же понимание дисфункции указывают авторы вышеупомянутой работы. Т.е., дисфункция в данном случае сродни гипофункции. На наш взгляд, общепринятый термин может означать и гиперфункцию. Но это вторично. Главный парадокс определения сепсиса заключается в следующем: морфологическая основа воспаления – гиперфункция клеток, составляющих органы и системы, должна сочетаться с гипофункцией или, даже, с недостаточностью, этих органов и систем.
Однако, главное, что заложено в данном понимании сепсиса, это клинически выраженная ПОН: не бактериемия или септицемия, а состояние организма, вернее его ответной реакции, которое характеризуется недостаточностью, т.е., срывом процесса адаптации.
Таким образом, принятый в настоящее время синдромный подход к пониманию сепсиса вносит свою лепту в диагностику тяжести состояния пациента, однако, на летальность пострадавших он, к сожалению, не повлиял. Хочется надеяться, что этиопатогенетическая сущность его трактовки все-таки будет приоритетной.
Анализ литературных данных показывает, что большинство исследователей считает системный воспалительный ответ (СВО) главной причиной развития жизне-опасных осложнений у пациентов с тяжелыми ожогами (Визир В.А., Березин А.Е., 2001;Лазанович В.А., Смирнов Г.А., 2001; Wang S et al, 2001; Schwacha MG, 2002, 2003; Kowal-Vernj A et al, 2005; Sakallioglu AE et al, 2006; Reyes R et al, 2006;Finnerty CC,2006; Козлов В.А., 2006; Hsing CH et al, 2007; Ipaktchi K et al, 2007).
Поэтому общепринятые способы лечения базируются, в основном, на подавлении воспалительной реакции. Это – длительное проведение гиперволемии, гемодилюции, гепаринотерапии и седации; мощная детоксикация, использование больших доз глюкокортикоидов и т.д. Причем, в настоящее время наблюдается тенденция к усилению и расширению этого арсенала на всех уровнях, включая клеточно-молекулярный.
Т.е., мы действуем по схеме: степени тяжести травмы (тяжести состояния) ® воспаления ® противовоспалительной терапии = отсутствие ожидаемого эффекта у пациентов с критическими ожогами. И, если последние две составляющие формулы логичны, то первые две – не очевидны, ибо известный эффект дозозависимости работает до определенного значения, а супердозы вызывают ингибирование.
По современным представлениям, системный воспалительный ответ - стадийная активация клеток (нейтрофилов, моноцитов/макрофагов, лимфоцитов, тромбоцитов, эндотелиоцитов), продуцирующих цитокины и другие медиаторы и формирующих «цитокиновую сеть». При чрезмерной активации это приводит к генерализации воспаления с утратой защитной функции локального воспалительного очага и нарастанием эффектов системной альтерации (Козлов В.К., 2006).
Известно, что тяжелая термическая травма сопровождается выраженным метаболическим истощением и клеточным дефицитом, что резко снижает как энергетическое обеспечение воспалительной реакции, так и количество ее участников. Т.е., у тяжелообожженных пострадавших имеются все предпосылки к ингибированию СВО, которые, на наш взгляд, продолжают углубляться вплоть до финального исхода, характеризующегося стремительно нарастающей и абсолютно не корригируемой ПОН – материальной основой полной воспалительной анергии.
В этой связи мы задались вопросом о правомочности проведения столь мощной супрессии воспаления у пациентов с критическими ожогами.
В настоящее время спектр процессов, обуславливающих, по сложившимся традиционным представлениям, системное поражение, расширился до молекулярно-генетического. Основными негативными синдромами при этом считаются повышенный стероидный фон, гиперметаболизм, оксидативный стресс, гиперактивация фагоцитоза, цитокиновая агрессия, индукция апоптотической клеточной гибели, экспрессия воспалительных генов и т.д.
Никоим образом не умаляя степени участия вышеуказанных процессов в системном воспалении, мы, тем не менее, склонны рассматривать это участие как необходимую составляющую универсальной ответной реакции организма на повреждение, которая, мобилизуя организм и расходуя его резервы, способствует восстановлению основных гомеостатических параметров и кожного покрова. Увеличение тяжести травмы, приводящее к гиперактивации этих процессов, сопровождается усилением расхода резервов с постепенным их истощением, лежащим в основе следующей ответной реакции на продолжающееся усиление повреждения.
Таким образом, мы предполагаем, что, у пациентов с тяжелыми, а тем более критическими, ожогами наблюдается раннее угнетение ответной воспалительной реакции организма, требующее соответствующей коррекции.
Исследование ответной реакции организма представляется методологически сложной проблемой. Однако, опираясь на творческое наследие классиков медицины, посвятивших себя изучению роли организма в развитии патологии, мы облегчили нашу задачу.
В основе общей реакции организма на любое повреждение лежит понятие гомеостаза, предложенное У. Кенноном (1927) и постулированное более ста лет назад К. Бернаром (1878): о значении постоянства внутренней среды для сохранения жизни в условиях изменчивости окружающей среды. К. Бернар и великий физиолог И.М. Сеченов считали биохимический дисбаланс в организме первопричиной большинства болезней.
В современном понимании гомеостаз характеризуют, скорее, как способ сохранения структуры и функции системы. Устойчивость организма к воздействию внешних факторов достигается путем приспособительных реакций, названных И.П. Павловым «физиологической мерой». Выдающийся канадский ученый Г. Селье обозначил эти механизмы общим адаптационным синдромом (1936), выделив в их развитии три фазы: тревоги, резистентности и истощения.
Большое значение целесообразности включения универсальных механизмов приспособления придавали такие выдающиеся ученые, как И.В. Давыдовский (декомпенсация этих механизмов – в основе патологии), Д.С. Саркисов (важнейшая роль воспаления в развитии универсальной приспособительной реакции; адаптивная внутриклеточная регенерация), П.К. Анохин (теория функциональных систем), Н. Виннер (постоянство переменной). Исследованиями закономерностей ответной реакции организма продолжают заниматься и в настоящее время (Меерсон Ф.З., Волчегорский И.А., Скулачев В.П., Нефедов В.П. и т.д.).
Таким образом, ответная реакция организма, имея защитно-приспособительный характер, необходима и не может быть оценена однозначно, тем более как патологическая.
Благодаря Г. Селье, выявившему активацию гипофизарно-надпочечниковой системы в качестве пускового механизма в развитии универсальной ответной реакции, появилась возможность оценить ее интенсивность. Позже неспецифический ответ организма на любое предъявляемое ему требование он назвал стрессом, а общий адаптационный синдром (ОАС) – синдромом биологического стресса.
Большое практическое значение Селье придавал стадии истощения, демонстрирующей, что способность организма к приспособлению, или адаптационная энергия, не беспредельна. Он писал, что, если повреждение незначительно, реакция заканчивается, изменения восстанавливаются. Если патологический фактор чрезмерен или длителен, развивается истощение коры надпочечников и наступает гибель организма. При перенапряжении функции реакция может стать неадекватной и, превратившись из физиологической в патологическую, вызвать запредельное торможение.
Необходимо отметить противоречивость литературных данных касательно выраженности активации этой системы. Большинство авторов связывают повышение стрессовой активности с негативной катаболической перестройкой, мышечной и системной кахексией, ослаблением сопротивляемости к инфекции (Udelsman R et al, 1986; Donald RA et al, 1993; Ortega AE et al, 1996; Hart DW et al, 2002;Dolecek R et al, 2003; Jeschke MG et al, 2004, 2005; Deitch EA et al, 2006; Ipaktchi K et al, 2007). Другие исследователи сообщают об адреналовой недостаточности у пациентов критических состояний, в том числе при сепсисе различной этиологии, и рассматривают активацию гипоталамо-гипофиз-адреналовой системы как адаптацию к достижению гомеостаза (Briegel J, 2000; Beishuizen A, Thhijs LG, 2001; Shroeder S et al, 2001; Loisa P et al, 2002; Prigent H et al, 2004).
Цель исследования - изучение процесса адаптации к ожоговой травме и разработка рекомендаций по его коррекции. Для оценки ответной реакции организма, включающей эндокринно-метаболические и иммунные звенья, проведено комплексное исследование 442 пациентов с ожоговой болезнью, распределенных на три группы в соответствии с тяжестью травмы, определяемой с учетом индекса Франка (ИФ).
Все пострадавшие после получения термической травмы находились в состоянии шока. Оказание первичной квалифицированной помощи по выведению пациентов из критического шокового состояния осуществлялось по месту жительства или получения повреждения. По мере стабилизации основных гемодинамических параметров и после консультации специалистов ожогового центра Института хирургии им. А.В. Вишневского больные транспортировались в настоящее учреждение, где им оказывалась специализированная медицинская помощь. В настоящее исследование включены пациенты, находящиеся на лечении в 1990-2006гг.
Первую группу составили 139 стабильно-тяжелых больных с общей площадью (S) поражения <50% и ИФ≤90. При поступлении состояние их было тяжелым, обусловленным как острой ожоговой токсемией, так и отсутствием квалифицированной медицинской помощи во время транспортировки. В дальнейшем состояние их оценивалось как стабильно-тяжелое, без признаков выраженной полиорганной недостаточности и тяжелого сепсиса. Пациентам с целью детоксикации была продолжена интенсивная терапия в условиях ожогового отделения.
Вторую и третью группы составили тяжелообожженные пациенты, оценка тяжести поражения которых свидетельствовала о неблагоприятном прогнозе — ИФ >100, а течение ожоговой болезни осложнилось развитием клинически выраженной ПОН и тяжелого сепсиса. Всем пострадавшим этих групп проводилось длительное комплексное интенсивное лечение, включающее реанимационную поддержку. Причем, вторую группу составили выжившие больные с общей S поражения ≥50% и ИФ≥135±10 (n=183), а третью — аналогичные по тяжести травмы пострадавшие с ИФ≥130±15 (n=120), но имеющие в дальнейшем неблагоприятный исход в течение 2-3-х недель после ожога (n=28) или в более поздний период, через 1.5-2 мес после травмы (n=92). Анализы проводились с момента поступления (7-10 сут), и далее, с периодичностью в неделю.
Оценка глюкокортикоидной функции коры надпочечников осуществлялась путем определения суточной экскреции 17 ОКС методом микроколоночной хроматографии (Bradlow HL, 1968), c использованием реактивов фирмы «Biosystems». Тотальное нарушение процесса канальцевой реабсорбции при тяжелой термической травме обуславливает диагностическую ценность определения метаболитов в моче (Дедов И.И., 2000). Особое значение имеет гипераминоацидурия (повышение экскреции аминокислот), свидетельствующее о преобладании потерь белка. Определение азотистого баланса производили по содержанию и экскреции аминокислот нингидриновой реакцией, предложенной Moore S и Stein W (1948). Измерение спонтанной хемилюминесценции крови проводили по методу Lindena J (1987) на люминометре (Япония). Результаты выражали в приборных единицах (мВ) в пересчете на 10 мкл крови. Определение однонитевых разрывов ДНК осуществляли измерением флюоресцентного свечения комплексов щелочных гидролизатов ДНК с бромистым этидием (Thyerry D et al, 1985) в модификации Москалевой Е.Ю. (1998). Экспрессия мРНК цитокинов и факторов апоптоза методом ПЦР. Полимеразную цепную реакцию в реальном времени (РТ-ПЦР) проводили с использованием специфических праймеров к ДНК цитокинов (TNF-a, IL-6, 10) и факторов апоптоза (каспаза 8, Bcl 2, Bax и P53), а также ревертазы, b-актина, Tag-полимеразы (реактивы фирмы «Синтол») на отечественном анализаторе нуклеиновых кислот АНК – 32. Мониторинг накопления амплифицированного продукта осуществляли с помощью флюоресцентного сигнала. Иммунный статус исследовалиметодами турбодиметрии, хемилюминесценции, иммунодиффузии. Количество клеток и фенотип лимфоцитов, относящихся к различным популяциям, а также молекулярные маркеры активации, зрелости и функциональной специализации клеток анализировали методом иммунофлуоресценции с помощью моноклональных антител и проточного лазерного цитофлюориметра FACSCalibur фирмыBecton Dickinson в отделе активации иммунитета ГНЦ Институт иммунологии МЗ РФ. Статистическую обработку и расчет коэффициентов корреляции проводили с использованием пакета программ Статистика-6.
Результаты представляли как M±m, где M – среднее арифметическое, а m – стандартное отклонение. Достоверность различий между группами оценивали с использованием критерия Уилкоксона для связанных величин и U- теста или Стьюдента для несвязанных величин. Достоверным считали различие при p£0.05. Для определения корреляционных связей использовали коэффициент линейной корреляции Пирсона (r). При оценке показателей мы использовали коэффициент отклонения среднего значения показателя в анализируемой группе от среднего значения нормы – Km.
Результаты и обсуждение
Несмотря на сложившееся мнение о постоянно повышенном уровне стрессовых гормонов у пациентов с тяжелыми ожогами, нами получены результаты, свидетельствовавшие о различиях в выбросе кортизола между пациентами наблюдаемых групп на всем протяжении острого посттравматического периода. У выживших тяжелообожженных по сравнению с пострадавшими с фатальным исходом уже при поступлении имеелось более, чем 6-кратное достоверное (p<0.01) увеличение содержания кортикостероидов: Kmпервых — 1.9, а вторых – 0.27. В дальнейшем среди пациентов с неблагоприятным исходом продолжало нарастать число больных, у которых экскреция биологически активных метаболитов кортизола не определялась вовсе.
Однако у стабильно-тяжелых больных c аналогичной по тяжести травмой, но не имевших жизнеопасных осложнений в виде тяжелого сепсиса и выраженной ПОН, определялась еще более интенсивная стресс-реакция: 3-кратная от средней нормы и 10-кратная по сравнению с пациентами с фатальным прогнозом. Причем, постепенный ее спад до нормы к 25-30 сут (p< 0.05) совпадал с клинической стабилизацией состояния.
Поэтому именно их уровень интенсивности стресс-реакции должен служить ориентиром для благоприятного прогноза и оптимальным для создания толерантности к постоянным стрессорным нагрузкам, включая агрессивные методы лечения и диагностики. А ранний спад стрессовых гормонов (срыв адаптации), явившийся прогностически неблагоприятным, необходимо своевременно корригировать назначением кортикостероидов.
При сравнительном исследовании пациентов с фатальным исходом на фоне болюсного введения супердоз кортикостероидов и без него нами продемонстрировано, что использование супрафизиологичных доз кортизола пациентам в раннюю фазу острого
периода приводит к увеличению летальности и ее темпов в среднем на 10% по сравнению с погибшими без применения супердоз.По-видимому, применение такого рода терапии возможно как экстра-мера в критических ситуациях для поддержания гемодинамики.
Известно, что летальный исход наступает иногда стремительно, в ранние сроки после травмы (3-4 недели). Причиной его чаще всего является генерализация инфекции. Течение болезни развивается в таком случае по гипердинамическому типу с кортикостероидной гиперактивацией. В четырех наблюдениях определялось стойкое увеличение экскреции 17 ОКС (6-10 кратное) практически весь наблюдаемый период. Однако гибель пациентов наступала на фоне септического шока, т.е., на фоне адреналовой недостаточности. Аналогичная картина наблюдалась у больных с «болюсной» гормональной терапией, у которых непосредственно в день гибели не определялись ни 17ОКС, ни сывороточное железо, ни продукты пероксидации.
Выявленные различия в интенсивности стрессовой реакции должны вызывать соответствующие изменения в метаболическом ответе: адаптивное умеренное усиление, дезадаптивные сверхусиление или ингибирование. Что мы и наблюдали при оценке некоторых важных обменных процессов, при этом дезадаптивность реакций выступала как свидетельство недостаточности соответствующих органов и систем.
Послеожоговый гиперкатаболизм, инициируемый стрессовыми гормонами и цитокинами, приводит к развитию истощения, иммунодефицита и сепсиса (Davies JWL, 1970; Лифшиц Р.И., 1977; Рудовский В, 1980; Downey et al, 1986; Fong YM et al, 1991; Hart DWet al, 2000, 2002; Pereira C et al, 2005).
В то же время, это адаптивная реакция, направленная на получение дополнительных источников для синтеза глюкозы: из гликогенных аминокислот (АК), получаемых путем распада белка, преимущественно, мышечного (Зайчик А.Ш., Чурилов Л.П., 2000; Almendro V., et al, 2003;Wolf S.E., et al, 2003; Duan X., et al, 2006).
Исследование баланса азота АК – общепризнанный метод оценки белкового катаболизма. Умеренная степеньгиперкатаболизма определялась у стабильно тяжелых пациентов, проявившаяся незначительным повышением содержания АК в крови (гипераминоацидемия) и достаточно выраженной их экскрецией (Km=3), отражающей преобладание потерь белка и тяжесть травмы как таковой.
Выжившие тяжелообожженные продемонстрировали адаптивное возрастание гипераминоцидемии (Km = 1.57 при p=0.054) на фоне дезадаптивного, достоверно резкого усиления экскреции азота (Km=7 при p <0.05), обусловленного почечной дисфункцией, а именно — нарушением энергозависимой канальцевой реабсорбции АК.
У пострадавших с фатальным прогнозом выявлен срыв адаптации: достоверные гипоаминоацидемия (Km=0.67 при p <0.0001) и резкое усиление гипераминоацидурии (Km = 8.5 при p <0.001), свидетельствующие о прогрессировании почечной недостаточности. Таким образом, сочетание гипоаминоацидемии (0.5 средней нормы) и гипераминоацидурии (8.5 средней нормы) оказалосьпрогностически неблагоприятным.
Как известно, в норме гипераминоацидемия является мощным стимулом синтеза белка. Однако у пострадавших с неблагоприятным прогнозом наблюдалось отсутствие эффективности от заместительной терапии растворами АК, проявившееся кратковременным повышением содержания азота АК в крови и резким его снижением в дальнейшем.
Обусловлено это, в первую очередь, тем, что у пациентов с неблагоприятным прогнозом наблюдалось усиление экскреции АК (Km на 15 сут — 9.5) на фоне их парентерального введения. В то же время, у выживших тяжелообожженных пациентов гипераминоцидурия в процессе лечения достоверно снижалась к 25 сут по сравнению с 8-10 сут (p<0.01) и 15 сут (p<0.0001), что свидетельствовало о снижении у них катаболизма, улучшении усвоения и нормализации функции почек на фоне комплексного интенсивного лечения.
Мониторинг азотистого баланса в процессе белково-заместительной терапии позволяет своевременно осуществлять коррекцию объемов вводимых нутритивных смесей и средств с гепа- и нефропротекторным действием для обеспечения адекватного лечения у всех тяжелообожженных пациентов.
Полученная путем гиперкатаболического расщепления белка глюкоза идет на синтез макроэргов для энергообеспечения воспалительной реакции. Необходимо отметить, что митохондриальный синтез АТФ реализуется путем свободно-радикального механизма и избыток образующихся в этой ситуации свободных радикалов негативно действует на окружающие клетки, вызывая состояние оксидативного стресса. Вторым основным источником свободных радикалов являются активированные нейтрофилы. Система нейтрофильного продуцирования оксидантов предназначена для локализации очага воспаления, однако чрезвычайно реакционные радикалы проникают в окружающие неповрежденные ткани и реагируют с фосфолипидами мембран, сульфгидрильными группами белков и т.д. Активация этих процессов идет параллельно повреждению антиоксидантных механизмов. Возникающий дисбаланс в про- и антиокидантной системах, наблюдаемый при тяжелых повреждениях, в том числе, ожоговых, создает условия для возникновения оксидативного клеточного стресса.
В числе последствий активации свободно-радикальных механизмов – перекисное окисление липидов (ПОЛ) биологических мембран, направленное на адаптивное в условиях гиперметаболизма повышение клеточной проницаемости. Однако в связи с безудержной самоподдерживаемой инициацией ПОЛ происходит не только модификация клеточной стенки, но и ее разрыхление и повреждение, приводящие к повышению микроваскулярной проницаемости и системному поражению органов и тканей.
Адаптивная умеренная активация пероксидации как свидетельство усиления свободно-радикального синтеза макроэргов и повышения клеточной проницаемости в условиях гиперметаболизма определена у выживших тяжелообожженных пациентов по интегральному показателю процессов ПОЛ – содержанию ТБК-продуктов. И дезадаптивное ослабление (Km = 0.9), свидетельствующее о недостаточности системы биологического окисления у пациентов с фатальным исходом.
Однако, наиболее реакционный показатель ПОЛ, малоновый диальдегид (МДА), имел тенденцию к значительному повышению уровня у стабильно тяжелых больных, нуждающихся, по-видимому, в усилении антиоксидантной терапии, особенно актуальной для пациентов с термоингаляционной травмой, при которой выявлена тенденция к дезадаптивно резкому повышению содержания метаболита, обусловленному, по-видимому, паренхиматозным поражением легких.
Ингибирование стрессовой воспалительной реакции приводит к хронизации ответа организма с неизбежным истощением. Поэтому причинами относительного ослабления процессов ПОЛ в дальнейшем у выживших тяжелообожженных пациентов II и абсолютного – у пострадавших с поздним летальным исходом III группы могут быть, по крайней мере, две. Во-первых, увеличение длительности запроса на энергетические субстраты приводит к их потреблению и невозможности адекватного синтеза макроэргов и, как следствие, ослаблению продукции свободных радикалов. Во-вторых, уменьшение функциональной активности нейтрофилов как следствие хронического энергодефицита и как дальнейшая причина ослабления процессов ПОЛ.
В итоге развивается недостаточность в системе биологического окисления (дефицит оксидазно-оксигеназных ферментов, осуществляющих перенос электронов, разрушение липидного микроокружения митохондриально-микросомального комплекса)
и возникает необходимость в заместительной терапии переносчиков электронов и протонов (цитохромы), применении методов по электрохимическому и физическому окислению крови, клеточной терапии.
Таким образом, различия в интенсивности стрессовой реакции у пациентов с разной по тяжести травмой и исходом находят свое отражение в уровне метаболических реакций, направленных на энергетическое обеспечение воспаления.
Отражая кратность исследуемых показателей от средней нормы и располагая их в соответствии со стресс-запускающей или стресс-лимитирующей направленностью можно составить адаптограммы, позволяющие проводить опережающую диагностику нарушений адаптивных реакций и своевременную их коррекцию.
Структурное моделирование ответной реакции организма на термическую травму, предложенное нами в виде адаптограмм, наглядно отображает вектор направленности процессов, определяемый как результирующая двух противоположных тенденций: стресс-запускающей и стресс-лимитирующей. По результатам проведенных исследований мы определили три основных типа адаптограмм: нормо-, гипо- и гиперстресс. Адаптивным является нормостресс с умеренным (2-3-кратным) повышением интенсивности ответа, дезадаптивными – гипо- (угнетение) и гиперстресс (6-10 кратное усиление) интенсивности стрессовой реакции, характерные для пострадавших с неблагоприятным прогнозом. Оба этих типа неизбежно приводят к гибели пациента без соответствующей корригирующей терапии.
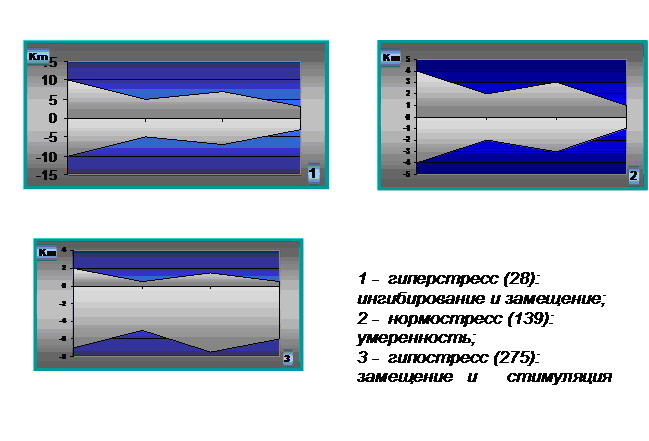
Рис.1 Основные типы адаптограмм при термической травме.
При этом главным критерием срочной адаптации будет постепенный спад стрессовой реакции на фоне относительного восстановления гомеостаза, а критерием срыва выступает преждевременное угнетение реакции стресс-адаптации на фоне выраженного метаболического истощения и сохраняющегося повреждения.
Выявление типа адаптограммы может способствовать более корректному ведению больных в остром периоде формирования срочной адаптации. Структурировать, отобразить графически можно любой анализ, что позволит оценить взаимоотношение двух противоположных систем.
Вершиной ответной реакции организма на повреждение является воспаление, так как именно благодаря ему происходит элиминация чужеродных антигенов, сначала посредством неспецифической реакции фагоцитоза (срочная адаптация), затем – специфической - антиген-антитело (долгосрочная адаптация).
Послеожоговая макрофагальная провоспалительная активация лежит в основе последующей иммунной дисфункции, присущей сепсису и ПОН, считают многие авторы. Однако известно, что острые гнойные инфекции сопровождаются как усилением фагоцитоза, так и его ингибированием, особенно, при генерализации инфекции, что ранее было продемонстрировано сотрудниками нашего института.
Выявленные отличия интенсивности эндокринно-метаболического ответа продолжают наблюдаться и при анализе первой стадии воспаления, процесса фагоцитоза, который оценивался по уровню спонтанной хемилюминесценции как показателю способности фагоцитов к продукции бактерицидных радикалов, индексу фагоцитоза как показателю задействования клеточных резервов и поглотительной функции фагоцитов и содержанию циркулирующих иммунных комплексов как функции неспецифического представления антигена.
Спонтанная продукция нейтрофилами бактерицидных радикалов, определяемая по интенсивности хемилюминесценции (СХЛ), при поступлении оказалась повышенной у выживших больных: в группе стабильно-тяжелых в среднем в 4.3, а тяжелообожженных – в 2.3 раза. При этом среднее значение уровня СХЛ в 1-й группе составило 869.83±240.44, а во 2-й – 462.70±48.64 ед., что свидетельствовало об активном функционировании циркулирующих нейтрофилов. В дальнейшем у стабильно-тяжелых пациентов гиперпродукция свободных радикалов на фоне проводимого интенсивного лечения постепенно снижалась, оставаясь однако умеренно-повышенной вплоть до 35 сут (Km = 2.2), отражая адаптивность воспалительной реакции.
Выжившие тяжелообожженные пациенты с относительным ослаблением свободно-радикальных процессов в первую неделю в дальнейшем, на протяжении всего срока наблюдения, имели тенденцию к их компенсаторному усилению, стабильно превышая в 3
раза среднюю норму и тем самым отражая запаздывание адаптивной реакции по активации фагоцитоза.
У пациентов с фатальным прогнозом в первую неделю после травмы наблюдалось резкое ослабление уровня СХЛ по сравнению с выжившими тяжелообожженными (p£0.05) и даже по сравнению с больными 1-й группы, у которых имелось относительное ослабление СХЛ во 2-ю неделю (p£0.05), что демонстрировало угнетение активности циркулирующих нейтрофилов по образованию бактерицидных свободных радикалов, которое может приводить к развитию сепсиса (Саркисов Д.С., 1987). Т.е. у данной группы пострадавших уже в первую неделю после травмы наблюдался срыв процесса адаптивного усиления фагоцитоза (Km = 0.9). В дальнейшем, к 3 и 4 недели, пострадавшие с неблагоприятным прогнозом так же, как и выжившие тяжелообожженные, имели достоверное повышение уровня СХЛ (p£0.05), обусловленное, по-видимому, стадией септикотоксемии. Однако эти пациенты так и не достигли степени бактерицидной активности, наблюдаемой у выживших больных.
Таким образом, при обширных и глубоких ожогах уже в первую неделю наблюдаются признаки ослабления фагоцитарных реакций вплоть до их ингибирования при фатальном прогнозе, что предполагает опережающее включение в комплексную терапию пациентов 2-й и, тем более, I3-й групп дополнительных мер по стимуляции бактерицидной активности нейтрофилов и окислительных процессов в печени.
Способность нейтрофилов к фагоцитозу у пострадавших с термической травмой была также усилена. Однако фагоцитарный индекс, определяемый как среднее число поглощенных частиц на нейтрофил, свидетельствовал о большей поглотительной способности у более тяжелых пациентов: в 1-й группе он был равен 3.14±0.75 и составлял 0.62 средней нормы, во 2-й - 5.16±0.51 и был несколько выше средней нормы (Km = 1.03). И максимальную поглотительную активность мы наблюдали у пострадавших с неблагоприятным в дальнейшем прогнозом. Индекс фагоцитоза у пациентов этой группы был повышен в 1.7 раза и составил 6.55±1.49.
В дальнейшем поглотительная способность нейтрофилов у пациентов с фатальным прогнозом 3-й группы резко падала, демонстрируя, продолжающееся ослабление фагоцитоза. Аналогичная дезадаптивность, но в меньшей степени, наблюдалась и у выживших с тяжелыми ожогами больных 2-й группы, в то время как стабильно-тяжелые пострадавшие 1-й группы имели возрастающую в этом отношении активность нейтрофилов при сохраненной способности к продукции бактерицидных радикалов.
Как проявление нарушения последовательности стадий фагоцитоза у выживших тяжелообожженных выступает обнаруженная нами обратная корреляция между СХЛ и индексом фагоцитоза слабой силы (r= -0.29), усиливающаяся к 15 сут (r= -0.44) и сохраняющаяся к 24 сут (r= -0.36).
Таким образом, более ранняя фаза процесса фагоцитоза – поглощение - исходно адаптивно повышается с тяжестью травмы, однако стадия киллинга, свидетельствующая о завершенности этого процесса и требующая, по-видимому, дальнейших энергетических затрат, демонстрирует прогрессивное ослабление фагоцитарной функции, особенно выраженное у пациентов с фатальным исходом. С учетом наиболее низкого уровня спонтанной хемилюминесценции при неблагоприятном прогнозе происходит перенасыщение нейтрофилов микробами с неспособностью к их перевариванию.
Образование ИК как адаптивная реакция предусматривает быстрое их выведение. В первую очередь, речь идет о макрофагах селезенки, где, главным образом и происходит деградация ИК. При нарастании количества ИК организм не может их вывести, и защитная адаптивная реакция превращается в патологическую.
Оценка уровня циркулирующих иммунных комплексов (ЦИК) демонстрирует усиление их образования у пациентов с термической травмой.
Причем, значения ЦИК отражали, по-видимому, активность предыдущего фагоцитарно-воспалительного процесса. Так, в первую неделю максимум содержания отмечался у стабильно-тяжелых больных 1-й группы (9.31±1.70), далее – выжившие тяжелообожженные 2-й группы (7.27±1.17) и, наконец, минимум определялся у пациентов с фатальным прогнозом (4.58±1.65). Различия между пациентами с тяжелыми ожогами 2-й и 3-й групп в эти сроки были достоверны (p£0.05).
В дальнейшем, пациенты с ожоговой болезнью средней степени тяжести в течение наблюдаемого периода практически элиминировали ЦИК, в то время, как у больных 2-й группы этот процесс задерживался, а 3-й – наоборот, развивался по восходящей, обуславливая дезадаптивность и этой реакции и наслоение негативных последствий накопления комплексов.
Таким образом, исходя из сути СВО как системного негативного последействия гиперфункции активированных клеток, в том числе, нейтрофилов, и на основании результатов эндокринно-метаболических и иммунных исследований мы можем констатировать наличие выраженного системного воспаления у стабильно-тяжелых пациентов с ИФ£90, его ослабление – у выживших пострадавших с ИФ³115 ±10 и резкое ингибирование — у больных с аналогичной по тяжести ожоговой травмой, но фатальным прогнозом.
В настоящее время известно, что глюкокортикоиды оказывают транскрипционный эффект, т. е., действуют на генетическом уровне: они активируют гены для хемокинов, цитокинов, семейства комплемента. Поэтому выявленные нами различия в протекании ответной реакции организма на термическую травму должны иметь логичные проявления на молекулярно-генетическом уровне.
Известно также, что индуцируемый повреждением оксидативный клеточный стресс вызывает повреждения ДНК, т.е. запускает апоптотическую гибель клеток. В норме эти разрывы инициируют репарацию, однако, количество их так велико, (превышает норму в
9-10 раз), что практически исключает возможность восстановления и через экспрессию гена ядерного белка p53 приводит к индукции апоптоза. По нашим данным при термической травме преобладают разрывы ДНК нейтрофилов, что естественно в условиях массового травматического поражения клеток и необходимости их фагоцитирования.
Выявленные ранее различия в проявлении интенсивности оксидативного стресса, совпадающие с типом протекания стрессовой реакции, нашли свое дальнейшее подтверждение и в результатах по определению разрывов ДНК: у крайне тяжелых больных с неблагоприятным прогнозом отмечается резкое снижение процента повреждений ДНК. Как же объяснить данное явление? В первую очередь, эффектом дозозависимости: крайне тяжелые ожоги вызывают массовую гибель клеток путем некроза, т.е., через грубое повреждение мембраны. Во-вторых, элементарным дефицитом клеток вследствие их потребления в процессе фагоцитоза. И, наконец, в условиях энергодефицита, организм не в состоянии утилизировать поврежденные клетки энергетически невыгодным апоптотическим путем.
С открытием молекулярной цитокиновой регуляции воспаления интерес к нему возобновился. И обусловлен он, прежде всего, новыми перспективами противовоспалительного воздействия на клеточно-молекулярном уровне.
Исходя из литературных данных о патогномоничности цитокинов при тяжелой термической травме, сопровождаемой ПОН и сепсисом, мы остановили свой выбор на прововоспалительных TNFa и IL-6, объединенных единой рецепторно-сигнальной связью с каспазой 8, и универсальном противовоспалительном IL-10, супрессирующем воспаление в целом, включая рост и дифференцировку клеток, но активирующем гуморальную фазу иммунного ответа. Мы также сочли необходимым исследовать экспрессию генов митохондриальных регуляторов программируемой клеточной гибели (апоптоза) белков семейства Bcl: проапоптотического Bax и противоапоптотического Bcl2. По мнению многих исследователей, от соотношения именно этих регуляторов зависит в конечном итоге реостат клеточной жизни или смерти.
Результаты наших дальнейших исследований цитокинового профиля и модуляции апоптоза методом РТ-ПЦР подтверждают литературные данные об инициации цитокинов и процесса апоптоза при термической травме.
Однако выявлено, что по мере утяжеления степени повреждения наблюдается не только усиление экспрессии мРНК провоспалительных TNFa и IL-6, но также достоверно, предположительно адаптивно, повышается экспрессия гена антивоспалительного IL-10, достигающая максимума на фоне минимальной провоспалительной активности у пострадавших с неблагоприятным в дальнейшем исходом, как в ранние (пог.II), так и в более поздние сроки (пог.I), что предполагает иную причину ингибирования – клеточную гибель.
При анализе активности митохондриальных регуляторов апоптоза нами обнаружена высокая их заинтересованность уже у стабильно тяжелых пациентов, демонстрирующая тяжесть ожоговой травмы как таковой и уровень борьбы за клетку. В то же время у пострадавших с ранним летальным исходом обнаружена преимущественно митохондриальная индукция апоптоза, в основе которой лежит, по-видимому, интрацеллюлярный энергетический коллапс.
У пострадавших с ИФ≤90 наблюдался провоспалительный цитокиновый фон (преобладание экспрессии IL-6, TNF-a над IL-10), индуцирующий рецепторно-каспазный путь апоптоза (9- кратное повышение активности каспазы 8) и наибольшая экспрессия генов митохондриальных факторов апоптоза (Bcl-2, Bax) при высокой активности ядерного p53. Причем, отмечается достаточно выраженная прямая корреляция p53 с Bcl-2: r=0.58 и слабая — с Bax: r=0.28, подтверждающая превалирование антиапоптотического регулирования.
Чтобы представить количественным образом про- или антивоспалительный и про- или антиапоптотический балансы, мы ввели соответствующие индексы: рецепторно-макрофагальный (РМ): TNFa+IL-6/IL-10, цитокин-воспалительный (Ц): IL-6/IL-10,митохондриальный (М): Bax/Bcl2 и апоптотический (A): Bax+p53/Bcl2. И поставили задачу оценить влияние тяжести травмы на эти показатели, т.е., на степень дисбаланса в исследуемых процессах.
Исходя из полученных нами данных у пациентов с ожоговой болезнью средней степени тяжести PМ = 9.47, Ц = 6.57, М = 0.9. Определив индексы по результатам только проб с экспрессией, мы более четко представим направленность выявленных отклонений: РМ = 11.2, Ц = 8.2, М = 1.3. Таким образом, используя предложенные индексы, можно конкретизировать предварительные выводы по данной группе больных, а именно, для них характерны: 1) выраженный провоспалительный цитокиновый фон; 2) индукция апоптоза по рецепторному и митохондриальному вариантам.
табл. 1
Экспрессия генов цитокинов и факторов апоптоза
у пациентов с ИФ ≤90
|
TNFa N 0.023±0.02 |
IL-6 N 0.02±0.01 |
IL-10 N 0.01±0.005 |
Cas-8 N 0.01±0.01 |
Bcl-2 N 0.01±0.005 |
Bax экспр. не выявлена |
p53 экспр. не выявлена |
|
0.061±0.03 (n=18) |
0.138±0.10 (n=18) |
0.021±0.01 (n=18) |
0.095±0.06 (n=18) |
0.359±0.16 (n=16) |
0.329±0.11 (n=16) |
0.536±0.20 (n=16) |
|
Экспрессия генов цитокинов и факторов апоптоза среди выявленных случаев
|
||||||
|
0.232±0.0 |
0.902±0.2 |
0.110±0.0 |
0.304±0.16 |
0.522±0.232 |
0.659±0.173 |
0.780±0.261 |
|
* 28% |
* 28% |
*28% |
* 28% |
*69% |
*50% |
*69% |
Примечание: результаты представлены в виде разности числа (M±m) критических циклов ПЦР.
* % выявленных случаев экспрессии от общего количества наблюдений (n).
При исследовании экспрессии mRNA в группе выживших тяжелых пациентов с ИФ ≥135±10 полученные результаты отражены в табл.2 аналогично предыдущим.
При анализе этих результатов обращает внимание максимальная экспрессия mRNA TNF-a и выраженная — у mRNA IL-6, 10 иBax, свидетельствующая об активация как рецепторной, так и митохондриальной инициации апоптоза; и минимальные экспрессииmRNA каспазы и p53. Видимо, при утяжелении степени травмы увеличивается количество поврежденных клеток, еще более активируя запуск к их апоптозу, но реализация его не может быть завершена, о чем свидетельствует угнетение каспазной активности у тяжелообожженных, лежащее в основе бактериемий.
При сравнении данных, полученных у пациентов двух групп, отмечается: 1) усиление экспрессии гена TNF-a у т/обожженных (p<0.05) на фоне появления слабой обратной корреляции с IL-6: r = (-0.22); 2) тенденция к общему повышению активности IL-6 и 10, свидетельствующая об усилении цитокинового ответа на травму, подкрепленная наличием между этими показателями прямой корреляционной связи выраженной силы: r = 0.8; 3) cнижение экспрессии гена p53 (p<0.05), регулятора клеточной гибели, и появление слабой обратной корреляционной связи с антиапоптотическим Bcl-2: r = (-0.26).
Более точные, на наш взгляд, тенденции могут выявить предложенные нами индексы. У тяжелообожженных выживших пациентов мы определяем: РМ – 3.7, Ц – 1.14, М – 2.4 и А – 2.9. По результатам проб с экспрессией: РМ – 2.7, Ц – 0.97, М – 1.5 и А – 2.0 (табл. 2). Т.е., несмотря на относительно высокую реализацию определяемых генов у данной группы пострадавших, становятся очевидны явления, связанные с утяжелением состояния пациента: ослабление активности макрофагов и воспаления (снижение РМ и Ц) и значительное поражение митохондрий (увеличение М), обуславливающее митохондриальную индукцию апоптоза (увеличение А) (рис. 2). Поэтому, несмотря на высокую активность как про-, так и противовоспалительных цитокинов, мы вынуждены констатировать у тяжелообожженных пациентов ослабление воспаления по сравнению с предыдущей группой. А по результатам проб с экспрессией выявляется даже тенденция к преобладанию антивоспалительного фона.
табл. 2
|
Экспрессии генов цитокинов и факторов апоптоза среди пациентов с ИФ ≥ 135±10
|
||||||
|
TNFa N 0.023±0.015 |
IL-6 N 0.02±0.01 |
IL-10 N 0.01±0.005 |
Cas-8 N 0.01±0.01 |
Bcl-2 N 0.01±0.005 |
Bax экспр. не выявлена |
P53 экспр. не выявлена |
|
1.164±0.480 (n=17) |
0.514±0.227 (n=17) |
0.450±0.246 (n=17) |
0.063±0.053 (n=17)
|
0.091±0.051 (n=16)
|
0.218±0.078 (n=16) |
0.048±0.022 (n=16) |
|
Экспрессия генов среди выявленных случаев |
||||||
|
2.197±0.767 |
1.247±0.424 |
1.275±0.581 |
0.268±0.211 |
0.292±0.131 |
0.435±0.112 |
0.154±0.042 |
|
*53% |
*41% |
*35% |
*23.5% |
*31% |
*50% |
*31% |
Примечание: результаты представлены в виде разности числа (M±m) критических циклов ПЦР.
* % выявленных случаев экспрессии от общего количества наблюдений (n).
С другой стороны, у этих же пациентов наблюдается повышение индексов митохондриальной инициации и реализации апоптоза. Причинами которых являются, по-видимому, прогрессирование клеточной гипоксии и митохондриальной проницаемости, приводящие к усилению дисбаланса белков семейства Bcl и, соответственно, к еще большей индукции апоптоза, а, значит, и клеточного дефицита, еще более ингибирующего воспалительный ответ. Порочный круг замыкается. Развивающиеся клеточный дефицит и иммуносупрессия, наблюдаемые у данной категории пациентов в большей степени, чем в предыдущей, являются абсолютными предпосылками к развитию ПОН и сепсиса.

Рис.2. Сравнительные значения цитокиновых индексов при термической травме
Данных по клиническому исследованию таких маркеров апоптоза, как митохондриальные пептиды семейства Bcl и ядерный белок p53, при термической травме или сепсисе мы не обнаружили.
Несомненную ясность в динамику показателей, выявленных у выживших пациентов с тяжелой термической травмой (ослабление воспаления, тенденция к антивоспалительному фону, митохондриальная индукция апоптоза и усиление гибели клеток) внесет анализ результатов исследования больных, погибших от аналогичной по тяжести травмы.
Среди пострадавших третьей группы выделены две подгруппы: 1) погибшие через 1.5 месяца и позже (поздняя гибель) и 2) с летальным исходом в первые 3-4 недели (ранняя гибель). Результаты их анализа представлены в таблицах 5-6.
Характерной особенностью данной подгруппы является антивоспалительный цитокиновый фон при поступлении, т.е. преимущественная экспрессия mRNA IL-10 по сравнению с IL-6 (p<0.02), а также ее возрастание по сравнению с пациентами «средней» степени тяжести (p<0.001). Необходимо отметить, кроме того, тенденцию к раннему усилению активности каспазы 8 по сравнению с обеими группами, как в уровне экспрессии, так и в проценте выявленных случаев. В то время как у пациентов I группы она составляла 28%, в группе выживших тяжелообожженных всего 23.5%, у больных с неблагоприятным в дальнейшем прогнозом отмечено усиление экспрессии mRNA cas 8 в 41% случаев. Возможной причиной последнего факта может быть реакция усиления индукции апоптоза на состоявшееся раннее системное инфицирование, которое, наслоившись на ответную реакцию организма на тяжелое повреждение, привело к истощению воспалительного цитокинового фона, оставив протеолитический след. При этом должны активироваться и другие маркеры, в частности, TNF-a. Однако зарегистрировано достоверное снижение экспрессии этого цитокина у пациентов с фатальным исходом по сравнению с благоприятным (p<0.01). Другой возможной причиной усиления каспазной активности могут быть, на наш взгляд, состояния гемодилюции и гипокоагуляции, усиливающие протеолиз и значительно осложняющие межклеточные рецепторные взаимодействия.
В дальнейшем, перед гибелью, у пациентов с неблагоприятным прогнозом наблюдается выраженное угнетение цитокиновой активности: достоверное снижение экспрессии mRNA TNF-a по сравнению с выжившими тяжелообожженными (p<0.05), нулевая экспрессия mRNA IL-6, -10.
Еще более прогностически значимыми в данной группе оказались определяемые индексы: резкое снижение как рецепторно-макрофагального (0.7), так и цитокинового (0.31) – при поступлении и нулевые значения – перед гибелью, демонстрирующие полное угнетение фагоцитарно-воспалительных реакций (табл. 3 и рис. 2).
Причинами ингибирования цитокиновой активности при поступлении является, по-видимому, супрессия антивоспалительным IL-10 и нарастающий дефицит клеток во все сроки ожоговой болезни, особенно, перед гибелью. Кроме того, необходимо иметь в виду, что проводимая интенсивная терапия также выступает в роли модулятора клеточной гибели.
табл. 3
Экспрессия генов цитокинов и факторов апоптоза
при поздней гибели пациентов с ИФ ≥ 130±15
|
При поступлении (до 7сут) |
Перед гибелью |
||||||
|
Tnfa |
IL-6
|
IL-10
|
Cas-8
|
Tnfa |
IL-6
|
IL-10
|
Cas-8
|
|
0.269±0.080 (n=17)
|
0.219±0.144 (n=17)
|
0.695±0.194 (n=17) |
0.124±0.052 (n=17) |
0.066±0.028 (n=12) |
0 (n=12) |
Следы (n=12) |
0.059±0.038 (n=12) |
|
Экспрессия генов среди выявленных случаев |
|||||||
|
0.508±0.095
|
1.243±0.549
|
1.312±0.201
|
0.301±0.093
|
0.198±0.014 |
0 |
Следы |
0.142±0.08 |
|
*53% |
*17.6% |
*53% |
*41% |
*16.6% |
*25% |
||
Примечание: результаты представлены в виде разности числа (M±m) критических циклов ПЦР.
* % выявленных случаев экспрессии от общего количества наблюдений (n).
Несомненную важность с позиций патогенеза и возможной терапии представляет сравнение показателей у пациентов с разными формами гибели. Для раннего летального исхода (табл.4) также характерен антивоспалительный цитокиновый профиль, причем абсолютный, при нулевой экспрессии mRNA IL-6.
На этом фоне резко выражена активность TNF-a и Bax (у 6 из 7 погибших), демонстрирующая усиление инициации рецепторного и митохондриального апоптоза. Достоверно усиление экспрессии Bax по сравнению с Bcl-2 (p<0.05). При сравнении с пациентами перед «поздней гибелью» достоверно повышение активности TNF-a (p<0.05), IL-10 (p<0.05). Достоверно также усиление экспрессии Bax у погибших в ранние сроки по сравнению с выжившими тяжелообожженными (p<0.05). Наблюдается тенденция к дальнейшему повышению активности каспазы 8 по сравнению со всеми предыдущими группами, которая в комбинации с высокой экспрессией TNF-a и Bax свидетельствует о выраженной инициации апоптотической гибели клеток. Определяемые индексы, результирующие баланс противоположных составляющих, также отражают ингибирование фагоцитарно-воспалительной реакции (РМ-0.57, Ц-0) и резкое увеличение митохондриального (12.4) и апоптотического (17.6) индексов (табл.4. и рис. 2).
Таким образом, для летального исхода в ранние сроки характерна резкая индукция апоптоза (митохондриальная инициация) на фоне полной иммуносупрессии, приводящая к стремительному распаду организма. В то время как у погибших в более поздние срокинаблюдается угнетение апоптоза также на фоне иммуносупрессии, следствием чего является развитие хронического воспаления, которое в условиях интенсивной детоксикации вызывает более медленный, но необратимый процесс клеточной гибели путем некроза.
Нарушения регуляции процесса апоптоза ядерным p53 также нашло свое отражение в динамике экспрессии mRNA этого белка при разной степени травмы и исходах. Максимум зарегистрирован в «средней» группе (0.536±0.200) и в подгруппе с ранним фатальным исходом (0.490±0.214). При этом, как уже сказано выше, его экспрессия имеет противоположную направленность: в первом случае – анти-, а во втором – проапоптотическую. Минимальная активность отмечена у тяжелообожженных выживших пациентов (0.048±0.022), изменение которой достоверно по отношению как к I группе (p<0.05), так и к последней подгруппе (p<0.05). Исходя из представлений об адаптивности ответной реакции организма можно предположить, что на данном примере четко прослеживается эволюция этого ответа: адаптивность в I, дезадаптивность – во II и срыв – в III группах.
|
табл.4 Экспрессия генов цитокинов и факторов апоптоза среди пациентов с ИФ ≥ 130±15, погибших в первые 3-4 недели
|
||||||
|
Tnfa
|
IL-6
|
IL-10
|
cas-8
|
Bcl-2
|
Bax
|
p 53
|
|
0.404±0.116 (n=7)
|
0 (n=7) |
0.714±0.288 (n=7)
|
0.139±0.09 (n=7) |
0.094 ±0.062 (n=7) |
1.170±0.391 (n=7)
|
0.490±0.214 (n=7) |
|
Средние значения экспрессии генов среди выявленных случаев |
||||||
|
0.472±0.112
|
0 |
1.25±0.258
|
0.243±0.142
|
0.220±0.114
|
1.365±0.401
|
0.858±0.235
|
|
*85.7% |
*57% |
*57% |
*42.8% |
*85.7% |
*57% |
|
Примечание: результаты представлены в виде разности числа (M±m) критических циклов ПЦР.
* % выявленных случаев экспрессии от общего количества наблюдений (n).
Таким образом, клиническое восприятие тяжелообожженного пациента как пострадавшего с более выраженным системным воспалением не находит подтверждения и по результатам молекулярно-биохимического исследования, которые демонстрируют разнонаправленную цитокиновую модуляцию процесса апоптоза в зависимости от тяжести травмы.
При ожоговом поражении, определяемом ИФ£90, наблюдается провоспалительный цитокиновый фон, умеренная макрофагально-рецепторная индукция клеточной гибели. Соотношение митохондриальных регуляторов апоптоза в пользу противоапоптотического bcl2 предполагает аналогичную направленность экспрессии гена p53, цель которой - репарация клетки на фоне клеточной гипоксии и угнетения окислительного митохондриального фосфорилирования. Комплекс реакций, направленных на провоспалительную умеренную активность, обеспечивающую апоптоз фагоцитов, «отработанных» по элиминации патогенов и поврежденных аутоклеток, является адаптивным.
При увеличении тяжести травмы до ИФ>100 степень выраженности клеточной гипоксии и поражения митохондрий значительнее, что находит свое отражение в проапоптотическом соотношении митохондриальных белков, инициирующих более массовую гибель клеток.
Значительнее и непосредственно травматическое поражение тканей, обуславливающих исходную клеточную гибель. Дополнительное задействование провоспалительных клеточных резервов для усиления фагоцитирования функционально неполноценных клеток оказывается неэффективным (по результатам исследования способности фагоцитов к продукции бактерицидных радикалов).
В такой ситуации организм как саморегулируемая система индуцирует адаптивный противовоспалительный ответ, направленный на супрессию клеточной гибели.
С другой стороны, универсальный противоапоптотический IL-10, ингибируя рост и дифференцировку клеток, удлиняет клеточную дисфункцию и ускоряет ее гибель.
Поэтому, антивоспалительный цитокиновый профиль, резко выраженная митохондриальная инициация апоптоза и преждевременное угнетение экспрессии генов цитокинов и факторов апоптоза, максимально выраженные у тяжелообожженных пациентов, особенно с неблагоприятным прогнозом, являются дезадаптивными реакциями.
Практическая значимость полученных результатов: интенсивная детоксикация должна проводиться с учетом степени воспаления и нежелательных последствий фармакологической модуляции апоптоза, а цитокиновая терапия в будущем – с учетом цитокинового профиля.
Цитокины не только продуцируются активированными иммунными клетками, но и сами участвуют в процессе их роста, дифференцировки, активации и гибели. Поэтому распределение основных популяций лимфоцитов должно быть отражением цитокинового профиля.
Результаты исследования основных популяций лимфоцитов у 67 пациентов с тяжелой термической травмой соответствуют известным литературным данным касательно клеточного дефицита и иммуносупрессии.
Вместе с тем у пострадавших с разными степенью тяжести и исходом выявлены отличия. У стабильно тяжелых пациентов на фоне общего клеточного дефицита определены относительно наиболее высокие показатели воспаления (хелперной активации) и супрессии, держащиеся на одном уровне в течение всего периода наблюдения, с тенденцией к относительному преобладанию супрессорной активности к 25 сут. Сравнительно с ними у выживших тяжелообожженных при поступлении и в дальнейшем отмечалось ослабление воспаления, особенно выраженное к 15 сут (p<0.05). При этом в течение исследуемого периода наблюдалась тенденция к превалированию супрессорной активности. Отмечался волнообразный характер показателей с критическим падением количества клеток всех популяций к 15 сут. В- клеточный дефицит, более выраженный, чем у стабильно тяжелых, также имел тенденцию к усугублению к 15 сут.
Напряженность про- и антивоспалительных потенциалов проявляется и в данных по корреляции Т-активных с Т-хелперами (r= 0.97) и Т-супрессорами (r= 0.81) в первую неделю после травмы. К 25 сут корреляция активных лимфоцитов с супрессорной популяцией снижается (r= 0.52), а с хелперной остается высокой (r= 0.89).
У пациентов с фатальным исходом обнаружено еще большее ослабление воспаления при поступлении, имеющее, однако, тенденцию к росту, но не достигающего уровня предыдущих групп. Парадоксально, но на фоне ингибирования воспаления супрессия была максимальна и имела тенденцию также к значительному ослаблению к 21-25 сут сравнительно с пострадавшими I (p<0.005) и IIгрупп в эти же сроки. При поступлении у пострадавших с неблагоприятным прогнозом отмечен сравнимый с пациентами стабильно тяжелой группы дефицит B‑ клеток. Однако к 25 сут наблюдается критическое падение представителей всех популяций, в том числе и B- клеток, обуславливая состояние иммунного паралича. Реализацией цитокиновой регуляции является тип формируемого адаптивного иммунного ответа: доминирование макрофагальной активации (хелперный первого типа- Th1) или антителообразования (хелперный второго типа -Th2), имеющий колоссальное значение для положительного исхода болезни.
Th1 и Th2 ответы имеют различные механизмы утилизации антигенов микроорганизмов и чужеродных клеток. В первом случае происходит преимущественная стимуляция макрофагов к внутриклеточному киллингу микробов, во втором – экстраклеточному, с помощью антител-зависимого цитолиза, сопровождаемого секрецией эозинофилами и базофилами локальных медиаторов. При этом, если Th2 развивается преждевременно, до завершения процесса фагоцитоза, АТ не смогут уничтожить микробы, находящиеся внутри фагосомы, и они будут продолжать размножаться. К тому же АТ- зависимый цитолитический медиаторный эффект обернется против собственных клеток. В этом случае организм будет подвергаться двойной агрессии: экзо- и эндо-.
В дополнение к результатам исследования раннего иммунного ответа и цитокинового статуса, продемонстрировавшим угнетение фагоцитоза и противовоспалительный цитокиновый профиль у пострадавших с неблагоприятным прогнозом, данные анализа баланса основных популяций лимфоцитов по преобладанию иммуносупрессии и ослаблению хелперной активации в этой группе, свидетельствуют о преждевременном формировании у них гуморальной фазы воспалительного ответа уже при поступлении, т.е., на 7-10 сут.
У выживших тяжелообожженных пострадавших с относительной выраженностью указанных процессов имеется тенденция к формированию Th2 ответа в эти же сроки.
Умеренная активность фагоцитоза, провоспалительный цитокиновый статус и Т- хелперная активация, лежащие в основе воспаления, подтверждают формирование Th1 ответа у стабильно тяжелых пациентов. Постепенное нарастание супрессорной активности к 25 сут на фоне относительной пролиферации B-клеток свидетельствуют о своевременной подготовке к формированиюTh2 ответа.
Таким образом, комплексное исследование иммунного статуса с позиций оценки адаптивности процессов при термической травме позволяет выявить тип формируемого иммунного ответа и его зависимость от тяжести травмы.
Дополнительные исследования иммунного ответа на тяжелую ожоговую травму у 42 тяжелообожженных пострадавших с применением проточной цитофлуориметрии подтверждают правомочность нашей гипотезы о превалировании гуморальной фазы воспаления по мере утяжеления степени травмы (табл.5).
табл. 5
Основные проявления иммунной активности у пациентов с ИФ ≥ 135±10
|
воспаление |
супрессия |
резервы воспаления |
резервы супрессии |
цитолиз |
истощение |
| Лейкоциты | CD3+ T-клетки | акт. HLA-DR+ NK | Акт. HLA-DR+ CD8+Т | Эозинофилы | Эритроциты |
| Лимфоциты | CD4+ T-клетки | ФенотипCD3-16+56- NK | «наивные»CD8+CD28+T | цитол.CD8+, сод. перф. | Ср.сод.гемоглобина в эритр. |
| Моноциты | CD8+ T-клетки | ФенотипCD3-16+56+ NK | цитол.NK, сод. перф. | палочкояд. нейтрофилы | |
| B-клетки | хемилюмин. спонт. | Акт. HLA-DR+ CD3+Т | активность компл.CH50 | сегментояд. нейтрофилы | |
| Иммуноглобулин M | фагоц. индекс | Акт. HLA-DR+ CD4+Т | активность компл.EH50 | фенотип CD3-16-56+- NK | |
| Иммуноглобулин E | «наивные»CD4+CD28+T | CD8+CD28+T после акт. | |||
ЦИК (в 3% ПЭГ) |
CD4+CD28+T после акт. |
По мере истощения основных эффекторных популяций (CD3+, CD4+, CD8+ T – клеток) наблюдается дополнительное задействование воспалительных резервов, обладающих аутоиммунной цитолитической активностью (эозинофилы и перфорин содержащие клетки), повышается активность комплемента, превалируют иммуноглобулины аллергической направленности (М и, особенно, Е).
Все эти факторы обуславливают усиление процессов цитолиза при утяжелении степени травмы, являющегося, по-видимому, основным механизмом клеточной гибели. Несостоятельность адаптивной реакции воспаления приводит к другой адаптивной реакции — гуморальному ответу. АТ — зависимый цитолиз, наслаиваясь на состоявшуюся в процессе системного воспалительного ответа гибель клеток, приводит к еще большему ее усилению, оказывая негативное воздействие и на здоровые ткани.
В таком случае возникает необходимость пересмотра этиотанатогенеза с позиций интоксикации на позиции клеточной гибели, которая и обуславливает развитие ПОН со всеми вытекающими последствиями. Этот факт, а также иммуносупрессия и противовоспалительный цитокиновый профиль приводят к ингибированию воспаления у тяжелообожженных пациентов с ИФ>100.
Таким образом, на основании комплексного сравнительного исследования пациентов, различающихся по степени тяжести и исходу, и, используя принципы адаптивности в оценке ответной реакции организма на тяжелую термическую травму, мы пришли к выводу о стадийности этого ответа при неблагоприятном прогнозе (рис. 3, табл. 6).
СТАДИИ ОТВЕТНОЙ РЕАКЦИИ ОРГАНИЗМА НА ТЕРМИЧЕСКУЮ ТРАВМУ

Рис. 3. Схема ответной реакции организма на термическую травму
табл. 6
ЭНДОКРИННО-МЕТАБОЛИЧЕСКИЕ И ИММУННЫЕ ПРОЯВЛЕНИЯ
СТАДИЙ ОТВЕТНОЙ РЕАКЦИИ ОРГАНИЗМА
проявлениястадии |
Стероид-ная реакция |
Гипер-катабо-лизм |
Оксида- тивный стресс |
Актива- ция фагоцитоза |
цитокиновый профиль |
форма клеточной гибели |
преобладание сигнальной индукции |
тип иммунного ответа |
|
1. СВО |
+ |
+ |
+ |
+ |
провоспа- лительный
|
апоптоз |
рецепторная |
Th1 |
|
2. АНТИ — СВО |
¯ |
¯ |
¯ |
¯ |
противо-воспалительный
|
апоптоз, некроз |
митохонд- риальная |
Th2 |
|
3. АНЕРГИЯ |
- |
- |
- |
- |
- |
Некроз |
митохонд- риальная |
Th2®иммунный паралич
|
Адаптивная ответная реакция, заключающаяся в умеренно выраженном системном воспалительном ответе, приводит в условиях традиционного комплексного интенсивного лечения к достижению срочной адаптации и дальнейшему выздоровлению.
В случае дезадаптивности ответа, наблюдаемого, в основном, при прогностически неблагоприятной ожоговой травме, и заключающегося в ослаблении системного воспаления, происходит хронизация процесса, для которого характерны адреналовая недостаточность, гипометаболизм, угнетение фагоцитоза, противовоспалительный цитокиновый профиль и иммуносупрессия. Данные признаки выявляют противоположную системному воспалению стадию – анти-СВО.
Разработка лечебной тактики этой категории пациентов требует отхождения от общепринятой схемы комплексной детоксикации. По-видимому, основными принципами этой терапии должны стать следующие: ослабление супрессии воспаления; комплексное замещение и стимуляция; осторожный подход к использованию фармакологических средств, угнетающих метаболизм, ослабляющих клеточное взаимодействие и индуцирующих клеточную гибель.
При своевременной диагностике и соответствующей коррекции данная стадия обратима. В противном случае развивается необратимая воспалительная анергия.
Надежды на снижение высокой летальности среди пациентов с тяжелой термической травмой связаны, прежде всего, с мониторингом нарушений адаптивных реакций и глубины дисфункции жизненно-важных систем. Выявление стадии ответной реакции организма, отличной у пациентов с разной степенью повреждения и прогнозом, должно определять различную стратегию лечения.
Выводы
1. Системная воспалительная реакция, индуцированная стрессовыми гормонами и провоспалительными цитокинами в ответ на термическую травму, носит адаптивный характер. Тяжесть состояния тяжелообожженных пострадавших обусловлена нарушениями адаптивных реакций, степень которых зависит от тяжести травмы.
2. Течение стрессовой реакции у пострадавших, различных по тяжести травмы и исходу, имеет принципиальные отличия. У стабильно-тяжелых пациентов с ожогами менее 50% поверхности тела (ИФ£90) наблюдается адаптивная стрессовая реакция с тенденцией к гиперфункции: умеренные стероидный фон, гиперметаболизм и усиление процессов прооксидации. У тяжелообожженных пострадавших с ожогами ³50% поверхности тела (ИФ ³130) выявлены глюкокортикоидная недостаточность, гипометаболизм и угнетение процессов прооксидации, носящие относительный характер. При неблагоприятном прогнозе развиваются раннее ингибирование стрессовой реакции и угнетение метаболизма, имеющиеабсолютный характер.
3. На основе структурного моделирования стрессовой реакции (адаптограмм) определены три основных эндокринно-метаболических типа ответной реакции организма на термическую травму: адаптивный – нормостресс и дезадаптивные — гипо- и гиперстресс.
4. Основным критерием срочной адаптации на термическую травму является возвращение метаболических гомеостатических параметров по мере стихания стрессовой реакции. Критерием срыва процесса адаптации выступает ингибирование стрессовой реакции на фоне отсутствия достижения гомеостаза.
5. Молекулярно-биохимические исследования цитокинового профиля и факторов апоптоза повышают эффективность опережающей диагностики тяжести состояния пациента с тяжелой термической травмой, при которой адаптивными реакциями являются провоспалительный цитокиновый профиль и умереннная рецепторная индукция апоптоза (нормостресс), а дезадаптивными – противовоспалительный цитокиновый статус и митохондриальная инициация апоптоза (гипостресс). При раннем неблагоприятном исходе наблюдается ингибирование макрофагальной и резкое усиление митохондриальной индукции клеточной гибели (гиперстресс). Поздний летальный исход приводит к угнетению экспрессии генов цитокинов и факторов апоптоза. т.е. срыву адаптивных реакций (гипостресс ® анергия).
6. Иммунный статус стабильно-тяжелых пострадавших (ИФ£90) характеризует активация фагоцитоза (Th1 ответ) с постепенным формированием Th2 ответа, что является адаптивной реакцией (нормо- с тенденцией к гиперстрессу). У пациентов с более тяжелыми ожогами (ИФ ³130) наблюдается дезадаптивная реакция – относительное угнетение фагоцитарного и преждевременное формирование гуморального ответа, усиливающее клеточную гибель (гипостресс). При фатальном исходе наблюдается срыв адаптивных реакций – иммунный паралич (анергия).
7. Ответная реакция организма на тяжелую термическую травму может включать три стадии: адаптивное системное воспаление (СВО), дезадаптивную супрессию воспаления (анти-СВО) и срыв адаптации — воспалительную анергию. В основе неблагоприятного течения — усиление клеточной гибели. Эти стадии должны определять соответствующую терапию.
8. Клинико-лабораторная оценка больных с ожоговой патологией должна базироваться на принципах теории адаптации.
Список литературы
1. Структурные основы адаптации и компенсации нарушенных функций. Руководство под редакцией Д.С. Саркисова. – М.: Медицина. 1987; 448.
2. Алексеев А.А. Ожоговый сепсис: диагностика, профилактика, лечение. Дис. … д-ра мед. наук. – М., 1993; 233.
3. Крутиков М.Г. Инфекция у обожженных: этиология, патогенез, диагностика, профилактика и лечение. Дис. …д-ра мед. наук.– М., 2005; 371.
4. Смирнов С.В., Спиридонова Т.Г., Логинов Л.П. и соавт. Современный взгляд на причины летальности и пути ее снижения у больных с обширными ожогами. Новые медицинские технологии в лечении тяжелообожженных. Сб. тез. науч. конф. 1997; 4-9.
5. Артамонов Р.Г. Сепсис. Новая концепция патогенеза и лечения// http: //www.medic-21 vek.com/referats/ref 26.htm. – по материалам British Medical Journal.
6. Christ –Crain M and Muller B. Diagnostic and Prognostic Value of Hormokines as Biomarkers in Severe Infections. Yearbook of Intensive Care and Emergency Medicine. 2007; 22-31.
7. Визир В.А., Березин А.Е. Иммуновоспалительная активация как перспективная концептуальная модель формирования и прогрессирования сердечной недостаточности. http://www.therapy.zp.ua/sn/sn2/view/1.htm.
8. Лазанович В.А., Смирнов Г.А. Уровень сывороточных цитокинов у больных с синдромом полиорганной недостаточности.Медицинская иммунология. 2001; 3(2): 148.
9. Wang S, Xu W, Cao Q. The influence of stress inhibition on the plasma levels of LPS, pro-inflammatory and Th/Th2 citokines in severely scalded rats. Zhonghua Shao Shang Za Zhi. 2001; 17(3): 177-180.
10. Schwacha MG, Chaudry IH. The cellular basis of post-burn immunosupression: macrophages and mediators. Int J Mol Med. 2002; 10(3): 239-243.
11. Kowal-Vern A, Webster SD, Rasmasubban S, Casey L, Bauer K, Latensser BA, Rubin DB. Circulation endothelial cell levels correlate with proinflammatory cytokine increase in the acute phase of thermal injury. J Burn Care Rehabil. 2005; 26(5): 422-429.
12. Sakallioglu AE, Basaran O, Karakayali H, Ozdemir BH, Yucel M, Arat Z, Haberal M. Interactions of systemic immune response and local wound healing in different burn depths: an experimental study on rats. J Burn Care Res. 2006; 27(3): 357-366.
13. Reyes R Jr, Wu Y, Lai Q, and all. Early inflammatory response in rat brain after peripheral thermal injury. Neurosci Lett. 2006; 16, 407(1): 11-15.
14. Finnerty CC, Herndon DN, Przkora R, Pereira CT, Oliveira HM, Queiroz DM, Rocha AM, Jeschke MG. Cytokine expression profile over time in severely burned pediatric patients. Shock. 2006; 26 (1): 13-19.
15. Козлов В.К. Дисфункция иммунной системы в патогенезе сепсиса: возможности диагностики. Цитокины и воспаление.2006; 2(5): 15-29.
16. Hsing CH, Chiu CJ, Chang LY et al. IL-19 is involved in the pathogenesis of endodoxic shock. Shock. 2007; 13 (PubMed).
17. Селье Г. Очерки об адаптационном синдроме. - М.: Медицина. – 1960. – 254с. Селье Г. Стресс без дистресса//lib.ru/PSIHO/SELYE/distree.txt
18. Меерсон Ф.З. Адаптационная медицина: концепция долговременной адаптации. М.: Наука. - 1981. – 278с.
19. Волчегорский И.А., Долгушин И.И., Колесников О.Л., Цейликман В.Э. Экспериментальное моделирование и лабораторная оценка адаптивных функций организма. – Челябинск. — 2001. — 167с.
20. Скулачев В.П. Феноптоз: запрограммированная смерть организма// Биохимия. - 1999. -N64.
21. Нефедов В.П., Ясайтис А.А., Новосельцев В.Н. и др. Гомеостаз на различных уровнях организации биосистем// Новосибирск: Наука. Сиб. Отд. – 1991. – 232с.
22. Udelsman R, Ramp J, Galluci WT, Gordon A, Lipford E, Norton JA, Loriaux DL, Chrousos GP. Adaptation during surgical stress. A reevaluation of the role of glucocorticoids. J Clin Invest. 1986; 77(4): 1377-1381.
23. Donald RA, Perry EG, Wittert GA, Chapman M, Livesey JH, Ellis MJ, Evans MJ, Yandle T, Espiner EA. The plasma ACTH, AVP, CRH and catecholamine responses to conventional and laparoscopic cholecystectomy. Clin Endocrinol (Oxf). 1993; 38(6): 609-615.
24. Ortega AE, Peters JH, Incarbone R, Estrada L, Ehsan A, Kwan Y, Spencer CJ, Moore-Jeffries E, Kuchta K, Nicoloff JT. A prospective randomized comparison of the metabolic and stress hormonal responses of laparoscopic and open cholecystectomy. J Am Coll Surg. 1996; 183(3): 249-256.
25. Hart DW, MD, Wolf SE, MD, Herndon DN, MD, Chinkes DL, PhD, Lal SO, DO, Obeng MK, MD, Beauford RB, MD, Mlcak RP, RT. Energy Expenditure and Caloric Balance after Burn. Ann Surg. 2002: 235 (1): 152-161.
26. Dolecek R, Tymonova J, Adamkova M, Kadlcik M, Pohlidal A, Zavodna R. Endocrine changes after burns: the bone involvement. Acta Chir Plast. 2003; 45(3): 95-103.
27. Jeschke MG, Barrow RE, Herndon DN. Extended hypermetabolic response of the liver in severely burned pediatric patients. Arch Surg.2004; 139(6): 641-647.
28. Deitch EA, Ananthakrishnan P, Cohen DB, Xu da Z, Faceteova E, Hauser CJ. Neutrophil activation is modulated by sex hormones after trauma-hemorrhagic shock and burn injuries. Am J Physiol Heart Circ Physiol. 2006; 291(3): 1456-1465.
29. Ipaktchi K, Claassen L, Niederbichler AD et al. Topical attenuation of burn wound inflammatory signaling reduces thymic apoptosis and lymphoid organ inflammation. 12th Congress of the European Burns Association (EBA). Budapest, Hungary, 2007; 12.
30. Briegel J. Cortisol in critically ill patients with sepsis: physiologic functions and therapeutic implications. Wien Klin Wochenschr. 2000; 112(8): 341-352.
31. Beishuizen A, Thijs LG. Relative adrenal failure in intensive care: an indentifiable problem recuiring treatment? Best Pract Res Clin Endocrinol Metab. 2001; 15(4): 513-531.
32. Shroeder S, Wishers M, Klingmuller D, Hofer M et al. The hypothalamic-pituitary-adrenal axis of patients with severe sepsis: altered response to corticotrophin-releasing hormone// Crit Care Med. – 2001 Feb. – N 29(2). – P. 450-451.
33. Loisa P, Rinne T, Kaukinen S. Adrenocortical function and multiple organ failure in severe sepsis. Acta Anaesthesiol Scand. 2002; 469 (2): 145-151.
34. Prigent H, Maxime V, and Annane D. Science review: Mechanisms of impaired adrenal function in sepsis and molecular actions of glucocorticoids// Crit Care. – 2004. – N 8(4). – P. 243-252.
35. Bradlow HL// Steroides. – 1968. – N 11. – P. 265-272.
36. Thierry D, Rigaud O, Duranton I. Quantitative measurement of DNA strand breaks and repair in gamma-irradiated human leukocytes from normal and ataxia-teleangiestasia donors// Radiat. Res. – 1985. – Vol. 102. – N 3. – P. 347-358.
37. Москалева Е.Ю. Молекулярно-биохимические механизмы развития вторичных иммунодефицитных состояний при действии различных экологических факторов// Дисс. на соискание уч. степ. д.б.н. – Москва. – 1998.
38. Davies JW. Protein metabolism following injury// J Clin Pathol Suppl. – 1970. – N 4. – P.56-64.
39. Лифшиц Р.И. Некоторые итоги и перспективы исследований в патохимии ожоговой травмы// Вопросы биохимии ожоговой травмы: Науч. труды. – Челябинск, 1977. – С. 124-131.
40. Рудовский В., Назиловский В., Зиткевич В., Зинкевич К. Теория и практика лечения ожогов. – М.: Медицина. – 1980. – 376с.
41. Downey RS, Monafo WW, Karl IE, Matthews DE, Bier DM. Protein dynamics in skeletal muscle after trauma: local and systemic effects// Surgery. — 1986 Mar. – Vol.99, N 3. – P.265-274.
42. Fong YM, Minei JP, Marano MA, Moldawer LL, et al. Sceletal muscle amino acid and myofibrillar protein mRNA response to thermal injury and infection// Am J Physiol. – 1991 Sep. – Vol.261, N 3, pt.2. – P.536-542.
43. Pereira C, Murphy K, Jeschke M, Herndon DN. Post burn muscle wasting and the effects of treatments// J Biochem Cell Biol. – 2005 Oct. – N 37(10). – P. 1948-1961.
44. Зайчик А.Ш., Чурилов Л.П. Основы патохимии. – Элби, СПб. – 2000. – 688с.
45. Almendro V, Carbo N, Busquets S, Figueras M, Tessitore L, Lopez-Soriano FJ, Argiles JM. Sepsis induces DNA fragmentation in rat skeletal muscle// Eur Cytokine Netw. – 2003. – Vol.14, N 4. – P. 256-259.
46. Wolf SE, MD, Thomas SJ, MD, Dasu MR, PhD, and all. Improved Net Protein Balance, Lean Mass, and Gene Expression Changes With Oxandrolone Treatment in the Severely Burned// Ann Surg. – 2003 June. – N 237(6). – P. 801-811.
47. Duan X, Berthiaume F, Yarmush D, and Yarmush ML. Proteomic analysis of altered protein expression in skeletal muscle of rats in a hypermetabolic state induced by burn sepsis// Biochem J. – 2006 July. – Vol.397, N 1. - P. 149-158.
48. Pruitt BA, Goodwin CW, Vaugman GM et al. The metabolic problems of the burn patient// Acta Chir. Scand. Suppl. – 1985. – N 522. – P. 119-139.
49. Ерюхин И.А. Хирургические инфекции: новый уровень познания и новые проблемы// Инфекции в хирургии. – 2003. – Т. 1, N1. – С. 2-7.